Abstract
Antibody-based drugs and vaccines against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are being expedited through preclinical and clinical development. Data from the study of SARS-CoV and other respiratory viruses suggest that anti-SARS-CoV-2 antibodies could exacerbate COVID-19 through antibody-dependent enhancement (ADE). Previous respiratory syncytial virus and dengue virus vaccine studies revealed human clinical safety risks related to ADE, resulting in failed vaccine trials. Here, we describe key ADE mechanisms and discuss mitigation strategies for SARS-CoV-2 vaccines and therapies in development. We also outline recently published data to evaluate the risks and opportunities for antibody-based protection against SARS-CoV-2.Main
The emergence and rapid global spread of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), has resulted in substantial global morbidity and mortality along with widespread social and economic disruption. SARS-CoV-2 is a betacoronavirus closely related to SARS-CoV (with ~80% sequence identity), which caused the SARS outbreak in 2002. Its next closest human coronavirus relative is Middle East respiratory syndrome-related coronavirus (MERS-CoV; ~54% sequence identity), which caused Middle East respiratory syndrome in 2012 (refs. 1,2). SARS-CoV-2 is also genetically related to other endemic human coronaviruses that cause milder infections: HCoV-HKU1 (~52% sequence identity), HCoV-OC43 (~51%), HCoV-NL63 (~49%) and HCoV-229E (~48%)1. SARS-CoV-2 is even more closely related to coronaviruses identified in horseshoe bats, suggesting that horseshoe bats are the primary animal reservoir with a possible intermediate transmission event in pangolins3. Cellular entry of SARS-CoV-2 is mediated by the binding of the viral spike (S) protein to its cellular receptor, angiotensin-converting enzyme 2 (ACE2)4,5. Other host entry factors have been identified, including neuropilin-1 (refs. 6,7) and TMPRSS2, a transmembrane serine protease involved in S protein maturation4. The SARS-CoV-2 S protein consists of the S1 subunit, which contains the receptor binding domain (RBD), and the S2 subunit, which mediates membrane fusion for viral entry8. A major goal of vaccine and therapeutic development is to generate antibodies that prevent the entry of SARS-CoV-2 into cells by blocking either ACE2–RBD binding interactions or S-mediated membrane fusion. One potential hurdle for antibody-based vaccines and therapeutics is the risk of exacerbating COVID-19 severity via antibody-dependent enhancement (ADE). ADE can increase the severity of multiple viral infections, including other respiratory viruses such as respiratory syncytial virus (RSV)9,10 and measles11,12. ADE in respiratory infections is included in a broader category named enhanced respiratory disease (ERD), which also includes non-antibody-based mechanisms such as cytokine cascades and cell-mediated immunopathology (Box 1). ADE caused by enhanced viral replication has been observed for other viruses that infect macrophages, including dengue virus13,14 and feline infectious peritonitis virus (FIPV)15. Furthermore, ADE and ERD has been reported for SARS-CoV and MERS-CoV both in vitro and in vivo. The extent to which ADE contributes to COVID-19 immunopathology is being actively investigated. In this Perspective, we discuss the possible mechanisms of ADE in SARS-CoV-2 and outline several risk mitigation principles for vaccines and therapeutics. We also highlight which types of studies are likely to reveal the relevance of ADE in COVID-19 disease pathology and examine how the emerging data might influence clinical interventions.
Mechanisms of ADE
ADE has been documented to occur through two distinct mechanisms in viral infections: by enhanced antibody-mediated virus uptake into Fc gamma receptor IIa (FcγRIIa)-expressing phagocytic cells leading to increased viral infection and replication, or by excessive antibody Fc-mediated effector functions or immune complex formation causing enhanced inflammation and immunopathology (Fig. 1, Box 1). Both ADE pathways can occur when non-neutralizing antibodies or antibodies at sub-neutralizing levels bind to viral antigens without blocking or clearing infection. ADE can be measured in several ways, including in vitro assays (which are most common for the first mechanism involving FcγRIIa-mediated enhancement of infection in phagocytes), immunopathology or lung pathology. ADE via FcγRIIa-mediated endocytosis into phagocytic cells can be observed in vitro and has been extensively studied for macrophage-tropic viruses, including dengue virus in humans16 and FIPV in cats15. In this mechanism, non-neutralizing antibodies bind to the viral surface and traffic virions directly to macrophages, which then internalize the virions and become productively infected. Since many antibodies against different dengue serotypes are cross-reactive but non-neutralizing, secondary infections with heterologous strains can result in increased viral replication and more severe disease, leading to major safety risks as reported in a recent dengue vaccine trial13,14. In other vaccine studies, cats immunized against the FIPV S protein or passively infused with anti-FIPV antibodies had lower survival rates when challenged with FIPV compared to control groups17. Non-neutralizing antibodies, or antibodies at sub-neutralizing levels, enhanced entry into alveolar and peritoneal macrophages18, which were thought to disseminate infection and worsen disease outcome19.
Fig. 1: Two main ADE mechanisms in viral disease.
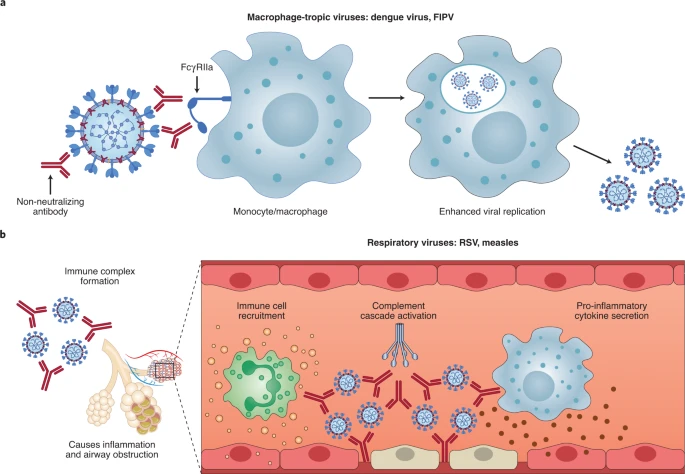
a, For macrophage-tropic viruses such as dengue virus and FIPV, non-neutralizing or sub-neutralizing antibodies cause increased viral infection of monocytes or macrophages via FcγRIIa-mediated endocytosis, resulting in more severe disease. b, For non-macrophage-tropic respiratory viruses such as RSV and measles, non-neutralizing antibodies can form immune complexes with viral antigens inside airway tissues, resulting in the secretion of pro-inflammatory cytokines, immune cell recruitment and activation of the complement cascade within lung tissue. The ensuing inflammation can lead to airway obstruction and can cause acute respiratory distress syndrome in severe cases. COVID-19 immunopathology studies are still ongoing and the latest available data suggest that human macrophage infection by SARS-CoV-2 is unproductive. Existing evidence suggests that immune complex formation, complement deposition and local immune activation present the most likely ADE mechanisms in COVID-19 immunopathology. Figure created using BioRender.com.
In the second described ADE mechanism that is best exemplified by respiratory pathogens, Fc-mediated antibody effector functions can enhance respiratory disease by initiating a powerful immune cascade that results in observable lung pathology20,21. Fc-mediated activation of local and circulating innate immune cells such as monocytes, macrophages, neutrophils, dendritic cells and natural killer cells can lead to dysregulated immune activation despite their potential effectiveness at clearing virus-infected cells and debris. For non-macrophage tropic respiratory viruses such as RSV and measles, non-neutralizing antibodies have been shown to induce ADE and ERD by forming immune complexes that deposit into airway tissues and activate cytokine and complement pathways, resulting in inflammation, airway obstruction and, in severe cases, leading to acute respiratory distress syndrome10,11,22,23. These prior observations of ADE with RSV and measles have many similarities to known COVID-19 clinical presentations. For example, over-activation of the complement cascade has been shown to contribute to inflammatory lung injury in COVID-19 and SARS24,25. Two recent studies found that S- and RBD-specific immunoglobulin G (IgG) antibodies in patients with COVID-19 have lower levels of fucosylation within their Fc domains26,27—a phenotype linked to higher affinity for FcγRIIIa, an activating Fc receptor (FcR) that mediates antibody-dependent cellular cytotoxicity. While this higher affinity can be beneficial in some cases via more vigorous FcγRIIIa-mediated effector functions28,29, non-neutralizing IgG antibodies against dengue virus that were afucosylated were associated with more severe disease outcomes30. Larsen et al. further show that S-specific IgG in patients with both COVID-19 and acute respiratory distress syndrome had lower levels of fucosylation compared to patients who had asymptomatic or mild infections26. Whether the lower levels of fucosylation of SARS-CoV-2-specific antibodies directly contributed to COVID-19 immunopathology remains to be determined. Importantly, SARS-CoV-2 has not been shown to productively infect macrophages31,32. Thus, available data suggest that the most probable ADE mechanism relevant to COVID-19 pathology is the formation of antibody–antigen immune complexes that leads to excessive activation of the immune cascade in lung tissue (Fig. 1).
Evidence of ADE in coronavirus infections in vitro
While ADE has been well documented in vitro for a number of viruses, including human immunodeficiency virus (HIV)33,34, Ebola35,36, influenza37 and flaviviruses38, the relevance of in vitro ADE for human coronaviruses remains less clear. Several studies have shown increased uptake of SARS-CoV and MERS-CoV virions into FcR-expressing monocytes or macrophages in vitro32,39,40,41,42. Yip et al. found enhanced uptake of SARS-CoV and S-expressing pseudoviruses into monocyte-derived macrophages mediated by FcγRIIa and anti-S serum antibodies32. Similarly, Wan et al. showed that a neutralizing monoclonal antibody (mAb) against the RBD of MERS-CoV increased the uptake of virions into macrophages and various cell lines transfected with FcγRIIa39. However, the fact that antigen-specific antibodies drive phagocytic uptake is unsurprising, as monocytes and macrophages can mediate antibody-dependent phagocytosis via FcγRIIa for viral clearance, including for influenza43. Importantly, macrophages in infected mice contributed to antibody-mediated clearance of SARS-CoV44. While MERS-CoV has been found to productively infect macrophages45, SARS-CoV infection of macrophages is abortive and does not alter the pro-inflammatory cytokine gene expression profile after antibody-dependent uptake41,42. Findings to date argue against macrophages as productive hosts of SARS-CoV-2 infection31,32.
....................................